Overview
Introduction
Prequips is a Java-based, modular software platform for integration, visualization and analysis of complex proteomics mass spectrometry data sets. The software has a graphical user interface and provides access to data produced by data analysis pipelines such as the Trans-Proteomic Pipeline. It bridges the gap between data processing pipelines and high-level analysis tools used in Systems Biology.
{kind=link}
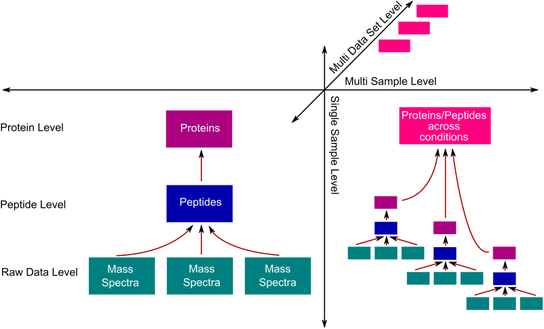
Spectral-, peptide- and protein-level data is loaded into the software straight from the files that are being produced by the data analysis pipelines (mzXML, pepXML and protXML). The software handles data sets consisting of several single and multi sample analyses, each of which can comprise several files. Analyses are assigned to projects, which is the highest level of organization. Prequips can handle several projects simultaneously.
All information within an analysis, i.e. spectrum-, peptide- and protein-level information is tightly integrated. In multi sample analyses the investigator can combine the information of several single sample analyses and perform higher-level analyses such as analysis of time series data. Prequips supports the Gaggle system to facilitate on-the-fly data exchange with third party applications.
{kind=link}
Click to enlarge | Prequips in the context of a data processing pipeline and high-level analysis tools.
Prequips provides a rich set of viewers to visualize information at all levels of an analysis. Viewers for raw data include spectrum and chromatogram viewers as well as a map-style viewer similar to Pep3D. Tabular viewers give an overview of all spectra, peptides and proteins associated with an analysis. Using these tools the investigator can start to explore the loaded data at any time and import additional data if required. Through the Gaggle the data can be mapped to and analyzed in the context of other types of relevant information such as interaction networks or gene expression data. Besides this Prequips has several options to export aggregated subsets of protein identifications and quantitations for use with external software.
Prequips is open-source software released under the GNU GPL. It is implemented in Java and based on the Eclipse Rich Client Platform. The modularity of Prequips enables labs to extended the software and adapt it to particular software environments. The use of Java renders the software virtually platform-independent.
Information Flow
Raw proteomics mass spectrometry data or spectral data is run through an analysis pipeline, which produces a set of heterogeneous output files at various stages of the data processing procedure. Prequips allows the investigator to import the output files produced by the tools in the data analysis pipeline and integrates the information across the spectral, peptide and protein level. Peptide and protein identifications can be enriched with information such as quality of the predictions or quantitative data for proteins and peptides.
Data Model and Integration
The basis for powerful visualizations and analyses is data integration. Prequips introduces two novel paradigms to handle heterogeneous data types and heterogeneous data sources. The first paradigm is concerned with how data is represented in Prequips and the second one is about the separation of the data sources from internal representations of the data.
Prequips has a sophisticated generic data model, which consists of two main parts: (1) core data and (2) meta information elements. Core data refers to all information that is commonly available in a proteomics mass spectrometry experiment such as spectra (as lists of peaks), peptide sequences and protein identifiers. Meta information elements represent data that describe the core data more closely. Every data analysis pipeline is creating such data at all stages and in various form. Examples for meta data are search scores dependent on the database search engine used, validation information produced by tools such as PeptideProphet or ProteinProphet, quantitation information by tools such as Libra or ASAPRatio.
{kind=link}
Extensibility with so-called data providers is the other key aspect of the data integration capabilities of Prequips. Data providers separate Prequips from the data sources and make the software independent of particular data formats. Plug-in interfaces have been designed for both core data providers and meta information element providers. Data providers for core data are implemented on either the spectrum, peptide or protein level. This separation with respect to analysis level is necessary for two reasons: (1) Data is currently stored separately for each level e.g. in mzXML, pepXML and protXML files and (2) it enables the investigator to load data from a particular level of interest independently of the other two.
Meta information element data providers work on existing analyses, that means before a meta information element provider can be used, core data structures must have been created. Meta information element providers read information from a data source and then map the information to the core data. How the target for a particular meta information element is identified depends on the target, for example a peptide can be identified by its sequence or its spectrum query identifier.
To address the problem of large data sets we have designed data providers that dynamically load data from the data source into memory when the data is being requested by the user. This approach allows Prequips to handle large data sets more efficiently and avoids long waiting times when a new data set is loaded. For instance, we have implemented a dynamic data provider for mzXML files that uses the index in those files to retrieve the list of peaks corresponding to a spectrum only when the investigator decides to visualize that spectrum. In this case only the index is being loaded when the file is first imported.
One of the main contributions of Prequips is the full vertical integration of spectral-, peptide- and protein-level information along with flexible integration of core data and meta information. As described before, the design of the software allows the investigator to load data for each level independently. Peptide-level information is required to establish a link between spectral- and protein-level information. Once peptide-level information has been imported Prequips will automatically map peptides to previously or subsequently loaded spectrum and protein information. The mapping is based on spectrum identifiers and peptide sequences to establish the link between spectra and peptides and proteins and peptides, respectively.